阅读:0
听报道

导语
《Nature》11月28日推出的一篇comments文章指出了使用高维数据的机器学习中常见的“坑”,以及避免方法,从而帮助该领域的小白能够客观的评价他们结果是否靠谱。方法论的文章会读起来较枯燥,因此本文尝试使用对话体来解读原文的3个主要问题。
论文题目:
Avoiding common pitfalls in machine learning omic data science
论文地址:
由于和生物有关的组学数据都是典型的高维数据,例如蛋白组,脂质组,转录组(RNA),基因组(DNA)等,在这个人工智能和医疗结合愈发深入的年代,基于高维数据的预测模型将会变得越来越重要。因此对于相关从业者,深入了解其中的方法论愈加不可或缺。另外,高维数据不止出现在生物相关的组学数据中,在材料,气象等领域也会有类似的数据集,故这篇“避坑指南”不仅仅适用于与生物相关的数据挖掘中。
过拟合与维度灾难
文章背景
东东:我先来讲一下这篇文章,有不对的地方请瑞瑞指出。这篇文章首先指出了过拟合和纬度诅咒问题。
瑞瑞:打断一下,在讲解文献时,要先说这篇文献的背景,就拿多组学数据来说,我们要解决的问题是什么,这些问题为啥之前没有出现?
东东:为了搞清楚那些因素影响我们的身体健康,人们对自身从多个角度进行了观测。随着测序成本的降低,从基因到转录出RNA再到合成蛋白质,积累了越来越多的数据。关于一个人的数据项,没有百万也有数十万,这其中的每一项数据,可以看成数据集的一个维度。而我们关心的是这些分子层面的数据如何与宏观的表型通过统计模型关联起来,例如血压,血糖,尿酸等数据变化与人体的健康状况的联系。
避免过拟合
瑞瑞:那在你刚才讲的故事中,过拟合意味着什么?
东东:过拟合指的是将表型之间本来是随机的变化错视为统计显著的关联,错误地和某一个维度建立了联系,即假阳性。
瑞瑞:那可以通过改变P值的预设范围来解决啊,比如之前都说P值小于0.05就是统计显著了,现在将P值变成0.01就好了,这样问题不就解决了?
东东:这样是不行的,因为假设有一百万维的数据,如果单独来看,那么就需要判定一百万次是否统计相关,而每次独立的判定假设有5%的几率将随机的误差当成是相关性的信号,那一百万次的判定,不知会导致多少次假阳性。
因此需要将P值的显著性判定值进行校正,最严格的就是用0.05除以数据的维度。但是由于不同数据间本身具有相关性,很多维度反映了身体相同的调控机制,所以简单地除以数据本身的维度,也会带来假阴性的问题。而且这些维度之间是有相互影响的。这两项原因,使得机器学习的方法逐渐流行起来。
瑞瑞:是的,最近有文献指出,在已发表的神经科学类论文中,有50%的文章统计学方法有疏漏[2],在其他的分子生物学领域也是类似的,毕竟该领域的数据暴增是最近十年间才发生的。
高维度低样本数据的维度诅咒(The curse of dimensionality)
瑞瑞:很多人不了解你之前说的维度诅咒指的是什么?对应于那种类型的机器学习任务?
东东:属于有监督学习。下图代表我们的数据集,其中有n个sample,比如有500个,但是每个人的基因数据,却可能有几万的维度,即p>>n,最上面的表型是不同颜色代表分类的标签,比如是否患某种疾病。但这里的基因数据是随机生成的高斯噪音。
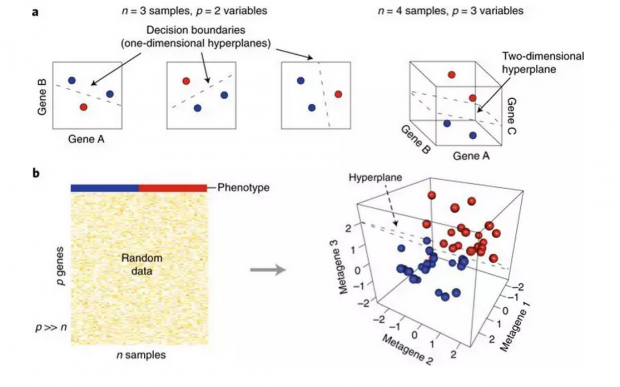
但是当使用SVM分类器,将原始投影在三维平面上时,却可以几乎完美地分开,这就是高维度低样本数据的维度诅咒(The curse of dimensionality)。
引入惩罚项的常规步骤
瑞瑞:机器学习中面对过拟合的常见方法,是引入惩罚项,如果模型越来越复杂,就在要优化的损失函数中加上对应的惩罚,这样是不是就能够解决问题了?
东东:惩罚项也有很多种类,该加那一类惩罚项这个问题也需要通过数据才能回答。
在理想情况下,有足够多的样本,能够将样本分成三部分,一个训练集,一个测试集,还有一个数据集用来确定模型的复杂度,这三个集合是完全隔离的;
先在训练集上使用不同的方法和惩罚项的组合训练一组模型及进行特征选择(选出哪些数据项对预测任务更有效);
之后在橙色的模型选择集合上选择一个最优模型,即图中曲线的最低点;
最后在测试集合上判定准确性。
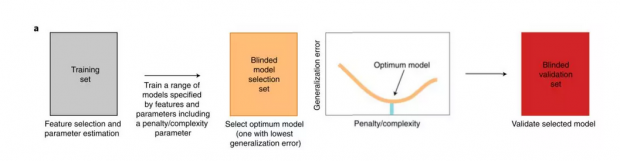
就像在下图中,先在测试数据集上训练了多个模型,之后在橙色的第三幅图中确定三次项的模型是最好的。最后再去测试数据集上看模型的泛化误差。
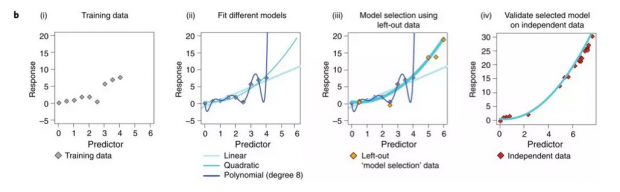
非理想情况下的数据集分类:交叉验证
瑞瑞:可是真实情况下,组学的数据往往本身就不会有那么多样本,不同标签间的比例也不一定均一,所以不能像理想中那样将数据集分成三类。那实际是怎样做的?
东东:类似下图的交叉验证,将数据分为N份,每次拿其中一份做测试集,剩下的做训练集。之后将每次实验验证集的误差汇总或取平均值,当做模型的泛化误差。由于每一份数据在都有机会被用做了验证集,因此对模型泛化误差的估计是无偏的。而模型选择的过程则是通过在不同模型上进行交叉验证完成的。
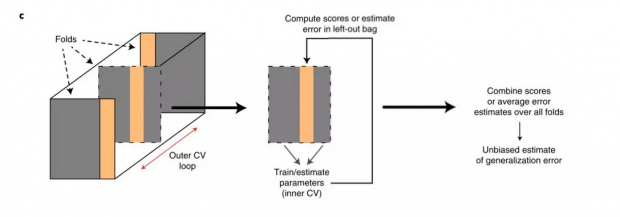
瑞瑞:是的,极端情况下,n是数据集的样本个数(leave-one-out CV ),即只拿出一个样本来,剩下的都用来做训练集,从而保证模型有足够的样本量。那在做交叉验证的时候,需要注意什么了?
东东:交叉验证的目的是为了避免训练出的模型过拟合,因此可以训练一组模型,之后将这些模型给予不同数据维度的权重进行平均及排序,从中根据模型复杂度的惩罚项来选出多少项对预测结果影响最大的特征。或者通过选择不同数量的模型,确定一个最优的惩罚项,之后再用全部的数据来训练这个加上了预估的最优惩罚项的模型[3,4,5]。
瑞瑞:如果已经进行了特征选择,比如选出了我们关注的表型和这几百个基因最相关,那在做交叉验证的时候需要注意什么?
东东:要在全部的数据项上进行交叉验证。使用降维后的数据以及反复的特征选择,会带来偏差项的提高。因此要区分CV用来判定整个模型的泛化能力的交叉验证外层循环以及用来对具体这个模型参数调优的内部循环,从而将模型选择和模型优化分离,从而避免过拟合。
未知的混淆因素
数据维度不足有什么不良后果?
瑞瑞:数据量不足之外,生物相关的数据还受制于数据本身的维度不足的影响,比如你收集的数据不包括生活习惯,或者做实验时用到的试剂的批次等,但这却会对这个人是否患病有显著的影响,或影响数据本身的分布。这种情况具体是怎样的?
东东:如下图所示,本来的数据集中一个没有被记录下的特征,将其称为X,如图中灰色和黑色的那一列,而在对应的表型上,这个X变量并不是均匀分布的,这导致在交叉验证时,不论在测试集还是验证集上,黑色对应的哪一列用肉眼就能看出其很特殊,这导致模型学到的其实不是判定一个样本是红色还是蓝色这个预设的目标,反而“偷懒”去判定样本是灰色还是黑色。
这导致的结果是不管训练集本身的交叉验证还是测试集,其结果都不差,但到了完全不同的一份独立数据集上,模型的表现就差得和随机乱猜差不多了。见右下方的ROC曲线,ROC 接近0.5,就意味着模型完全没有预测效力。
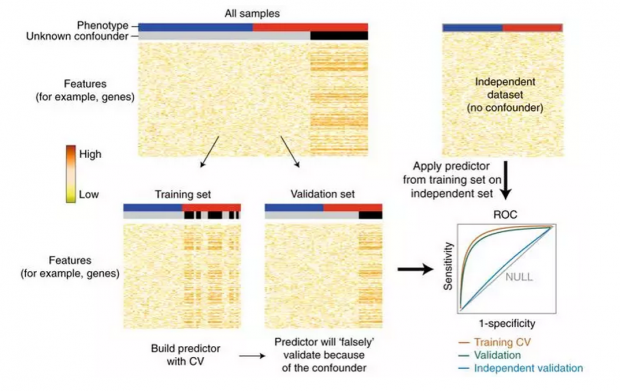
瑞瑞:因此当前判定一个模型是否靠谱的金标准,都是将你模型在其他实验室使用相同的实验方法对相近或相同样本观测得出的独立数据集上跑一下。通过一个独立的验证集,能够看到模型本身是不是受到未知的干扰因素的影响。那如果发现了有未知因素的影响,又应该怎么办呢?
对于未知混淆因素
东东:有些好排除,例如人种,年龄之间的差异,可以通过统计上的校正解决,对于年龄,由于年老对身体的影响不一定是线性而可能是指数的,因此还会将年龄的平方项和立方项作为控制因素[6]。
但更多的混合因素则是未知的,例如那一天做的实验,用的那一台机器等。虽然目前已有相关的统计方法来解决这一问题,但这些模型都假设未知的因素满足相应的分布,但现实中却往往不是这样的,这导致即使校正后也有残余的未知因素影响。
瑞瑞:当你的模型在独立数据集上无法重复好结果的时候,也不应该当成是你的模型的末日审判,而应该去找可能的原因和解释。
东东:比如用于验证的独立数据测量的数据项不如原始的用于交叉验证的数据项丰富,或者用来验证的数据来自一个不同的人种,从而使得你的模型不适用。尤其在医疗领域,为了保护隐私及伦理要求,并不是所有的数据都是公开的。这使得评价用于验证的独立数据集是否恰当变得很困难。
瑞瑞:这个问题对于那些方法类的“虐前任”型创新尤其严重。很多文章宣称自己通过整合很多组学数据,提出了一种比之前所有模型更准确的预测模型。但由于有残留的未知因素,当你引入新的数据项的时候,模型不做改进,就有可能效果比前人的好,这并不代表着你做出了方法学上的改进与创新。因此现在严谨的做法是如果你要证明你的方法有所长进,至少需要在五到六份的独立数据集上展示你的方法比前人的有显著提升。
非监督学习中也有过拟合
瑞瑞:在非监督学习中,会不会也出现过拟合的现象呢?
东东:有可能出现,例如在对特征进行聚类时,即使每一个数据项都和待研究的表型统计相关性并不显著,但它们之间的相关性却会使它们聚在一起,从而导致数据项聚成簇,但这样的结果却不能用于指导特征选择,否则就会导致过拟合。
瑞瑞:非监督学习的另一个常见应用是数据降维,这中间会不会也导致过拟合了?
东东:这篇论文中举出了一个具体的例子,如下图。图中的红色和绿色是两个样本在不同基因区域上的对应特征,红色的是有病的,绿色的是没病的。在图a中,使用数据的均值作为降维后的特征,会导致对数据分类时效果变差,但使用数据的方差则不会。而在b图中,由于数据本身的分布不同,导致相反的结果,使用方差会导致分类时效果变差。
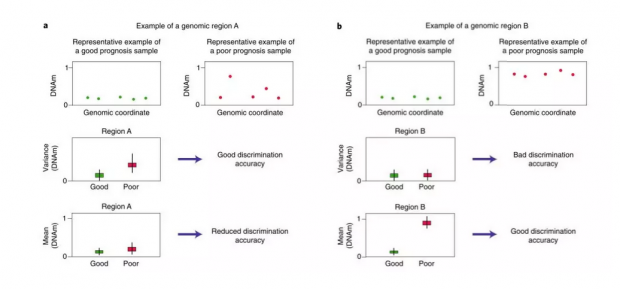
瑞瑞:虽然取方差和取均值不是现实中用到的数据降维的方式,但上面的例子展示了如果只根据少量数据选择的降维模型不适合新的数据,即导致过拟合。这方面有一个工具可以用来评估,可以进一步学习。

论文题目:
MOVIE: Multi-Omics VIsualization of Estimated contributions
论文地址:
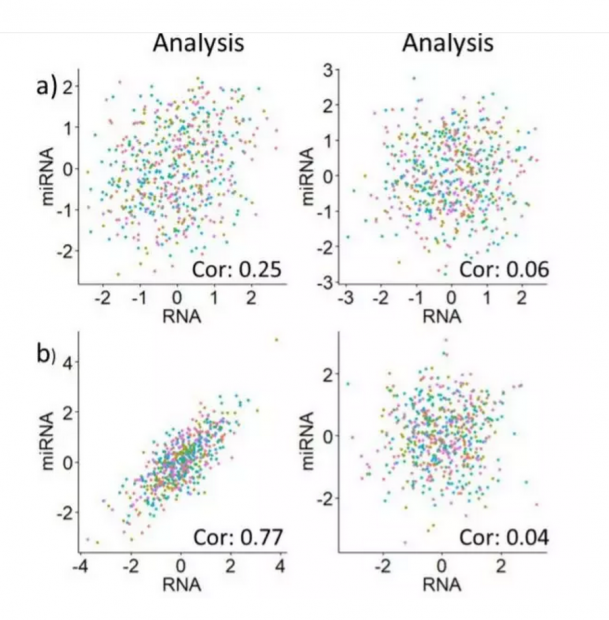
例如下面的图片中,哪一个过拟合了?
小结
这篇文章总结了高维数据的三个常见问题,一是没有用好交叉验证,导致预测模型过拟合。二是忽略了未知的干扰因素,导致模型在独立数据集上表现糟糕,三是在非监督学习中忽略了过拟合,导致特征选择时丢失关键信息,从而影响预测模型的效果。针对这三个问题,作者给出了当前行业内共识的常见解决建议,虽然这些问题都没有完全解决,但避免前人踩过的坑,是必不可少的。
参考文献
Nieuwenhuis, S., Forstmann, B. U. & Wagenmakers, E. J. Nat. Neurosci. 14, 1105–1107 (2011)
Simon, R., Radmacher, M. D., Dobbin, K. & McShane, L. M. J. Natl Cancer Inst. 95, 14–18 (2003)
Varma, S. & Simon, R. BMC Bioinform. 7, 91 (2006).
Teschendorf, A. E. et al. Genome Biol. 7, R101 (2006)

作者:郭瑞东
编辑:王怡蔺
话题:

0
推荐
财新博客版权声明:财新博客所发布文章及图片之版权属博主本人及/或相关权利人所有,未经博主及/或相关权利人单独授权,任何网站、平面媒体不得予以转载。财新网对相关媒体的网站信息内容转载授权并不包括财新博客的文章及图片。博客文章均为作者个人观点,不代表财新网的立场和观点。
 京公网安备 11010502034662号
京公网安备 11010502034662号 {kind=link}